
杜克大学的生物医学工程师开发了一个人工智能平台,可以自主比较分子,并从它们的变化中学习,以预测对发现新药至关重要的属性差异。该平台为研究人员提供了一个更准确、更有效的工具,以帮助设计治疗药物和其他具有有用特性的化学物质。
这项研究发表在10月27日的《化学信息学杂志》上。
机器学习算法越来越多地用于研究和预测药物开发和其他材料设计任务中使用的小分子的生物、化学和物理性质。这些工具可以帮助研究人员了解分子的关键“ADMET”特性——它是如何被吸收、分布、代谢、排泄的,以及它在体内的毒性。通过了解这些不同的特性,研究人员可以识别分子,从而开发出更安全、更有效的新疗法。
虽然现有的机器学习平台使研究人员能够筛选更多的分子,而不是在实验室中进行物理制造,但他们一次只能预测一个分子的性质,这限制了他们在确定最优化合物时的整体效率。
虽然有一些其他的计算方法可以省去这个额外的步骤,直接比较分子,但它们的范围有限。例如,像自由能摄动这样的方法是非常精确的,但由于计算复杂,它们一次只能评估少数分子。另一方面,像匹配分子对这样的方法要快得多,但只能比较非常相似的分子,限制了它们的广泛应用。
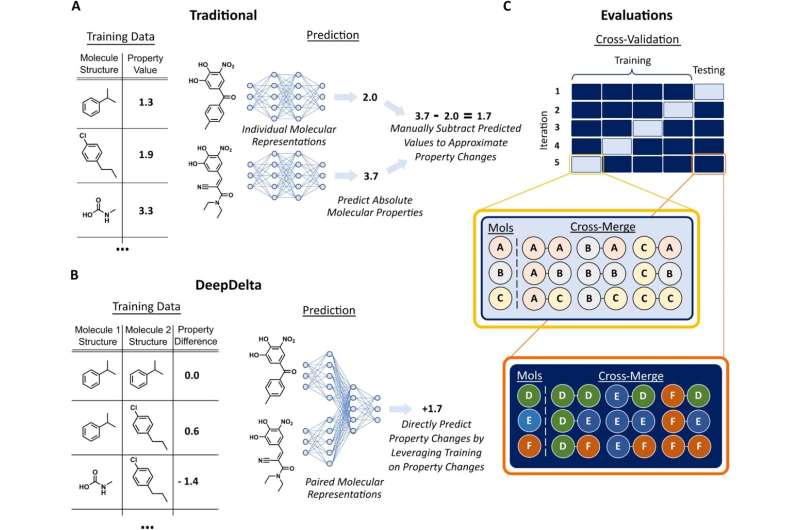
为了解决这个问题,Reker和Reker实验室的博士生Zachary Fralish开发了DeepDelta,这是一种深度学习方法,可以同时有效地比较两个分子,并预测它们之间的性质差异,即使它们非常不同。
Reker说:“通过让神经网络从一对一的比较中学习,你给它提供了比一次从一个分子中学习更多的数据点。”“该平台正在学习每个分子的结构和性质,但它也在学习两者之间的差异,以及这些差异如何影响分子的性质。”
该团队将DeepDelta平台与该领域最先进的两种模型进行了测试:Random Forest(一种广泛使用的经典机器学习模型)和ChemProp (DeepDelta基于的深度神经网络)。每个系统比较了两种已知的分子结构,并预测了10种不同的ADMET特性,包括分子如何从肾脏中清除,它们各自的半衰期以及它们在肝脏中的代谢情况。
事实证明,与现有平台相比,DeepDelta在预测和量化分子之间的分子特性差异方面更加有效和精确。
弗拉利什说:“对分子差异的训练使这种方法在确定一种新化学物质比现有化学物质更好或更差时更加准确。”“这就像做作业,更像是你的考试。我们还通过配对极大地扩展了数据集的规模,本质上给了我们的模型更多的作业,这确实有助于数据饥渴的神经网络学习更多。”
该团队现在期待着将该模型纳入他们的工作中,以设计潜在的新疗法并优化现有的候选药物。
弗拉利什说:“有了这个工具,我们可以研究一种几乎通过FDA批准的药物,但可能它有肝毒性的问题,所以它没有完全通过。”“DeepDelta可以帮助识别具有相同良好特性但没有肝毒性的分子。该工具通过帮助我们确定哪种化学品最有可能在现实世界中实现我们的目标,从而节省时间和金钱,从而开辟了许多机会。”




